This web page was created as an assignment for Genetics 677, an undergraduate course at UW-Madison.
Background

Hutchinson-Gilford Progeria
Hutchinson-Gilford Progeria Syndrome, or HGPS, is a rare genetic disorder with only about 100 documented cases in scientific literature. The disease is characterized by accelerated aging beginning at 6-12 months of age and, on average, is fatal by age 13 (1). Clinical findings show progressive progeroid features including scleroderma, alopecia, and limited bodily growth (3). de novo point mutations in the LMNA have been identified as the cause of nearly all HGPS cases (4). There has been one case identified involving an Indian family in which there was autosomal recessive inheritance of HGPS, however, that appears to be an exception as it is the only such documented case (2). Currently there is no cure for this disease, with common treatment focusing on maintaining cardiovascular health with low doses of aspirin (1). Recently phase II clinical trials have begun using the anticancer drug, Lonafarnib, which is an Farnesyltransferase inhibitor (FTIs) (5). In both mouse models studied thus far, disease phenotypes were improved by inhibiting an accumulation of farnesyl-prelamin A in the cells, which is the molecular basis of progeroid symptoms (5).
LMNA Gene
de novo mutations in the LMNA gene have been identified as the cause of Hutchinson-Gilford Progeria Syndrome. The LMNA gene codes for two proteins, lamin A and C, and is located on chromosome 1q (4). In most HGPS cases there is a single base mutation at position G608G within exon 11, with a cytosine being replaced with a thymine (4). This causes the activation of a cryptic splice site within the exon and leads to the synthesis of an abnormal lamin A protein that lacks 50 amino acids near the carboxy terminus . The mutated lamin A protein causes visible abnormalities in the nuclear envelope of the cells. This disease intrigues scientists as it may offer a great deal of insight into the human aging process, also there has been significant progress in the past several years towards finding a cure for HGPS. I would like to examine the genetics behind this disease and also document the ongoing progress in this area of medical genetics.
LMNA Gene Mutation
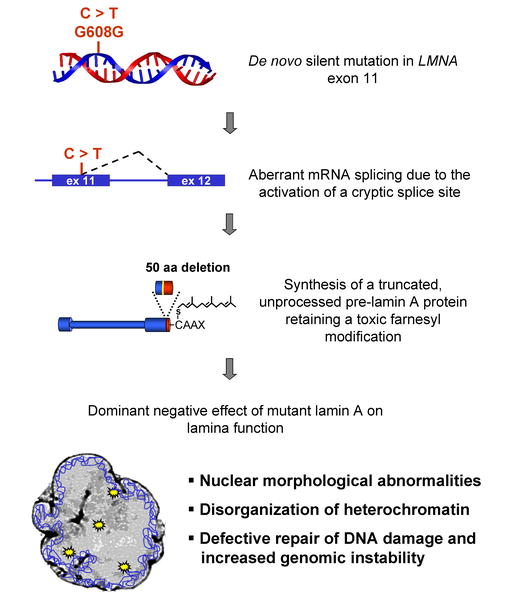
Fig. 1 de novo mutation in exon 11 of LMNA gene accounting for molecular basis of HGPS.
References
1. http://www.mayoclinic.com/health/progeria/DS00936.
2. Plasilova, M., Chattopadhyay, C., Pal, P., Schaub, N. A., Buechner, S. A., Mueller, Hj, Miny, P., Ghosh, A., & Heinimann, K. (2004). Homozygous missense mutation in the lamin A/C gene causes autosomal recessive Hutchinson-Gilford progeria syndrome. Journal of Medical Genetics, 41, 609-614. doi:10.1136/jmg.2004.019661
3. Sagelius, H., Rosengardten, Y., Hanif, M., Erdos, M., Rozell, B., Collins, F., & Eriksson, M. (2008). Targeted transgenic expression of the mutation causing Hutchinson-Gilford progeria syndrome leads to proliferative and degenerative epidermal disease. Journal of Cell Science, 121, 969-978. doi: 10.1242/jcs.022913
4. Eriksson, M., Brown, W. T., Gordon, L. B., Glynn, M. W., Singer, J., Scott, L., Erdos, M. R., Robbins, C. M., Moses, T. Y., Berglund, P., Dutra, A., Pak, E., Durkin, S., Csoka, A. B., Boehnke, M., Glover, T. W., & Collins, F. S. (2003). Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature, 423, 293-298. doi: 10.1038/nature01629
5. Meta, M., Yang, S. H., Bergo, M. O., Fong, L. G., & Young, S. G. (2006). Protein farnesyltransferase inhibitors and progeria. Trends in Molecular Medicine, 12(10), 480-487. doi: 10.1016/j.molmed.2006.08.006
6. (Figure 1). Scaffidi P, Gordon L, Misteli T (2005) The Cell Nucleus and Aging: Tantalizing Clues and Hopeful Promises. PLoS Biol 3(11): e395. doi:10.1371/journal.pbio.0030395
7. Image of child taken from maldova.org
Peter St. Andre [email protected] Last updated:2/3/09
gen677.weebly.com
