This web page was created as an assignment for Genetics 677, an undergraduate course at UW-Madison.
Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome
Erickson et al.
In May 2003 Erickson et al. published an article in Nature describing their findings of a de novo mutation leading to Hutchinson-Gilford progeria syndrome (HGPS). HGPS is an accelerated aging disease that is lethal, on average, by the age of 13 due to heart disease in 90% of the cases. At the time of research for this particular paper, the leading hypothesis for the cause of HGPS was sporadic autosomal dominant inheritance. The researchers for this paper felt there was the possibility of autosomal recessive inheritance of this disease. With this idea in mind the team ran a genome-wide scan searching for evidence of homozygosity.
DNA samples from 12 classical HGPS subjects were analyzed using 403 polymorphic microsatellite markers with an average spacing of 9.2 cM. Two of the samples indicated uniparental isodisomy (UPD) of chromsome 1q. This finding was out of the ordinary after further testing and possible causes for this will be discussed later. Previous reports discussed karyotype abnormalities in a monozygotic twin with HGPS. A mosaic cell population containing 70% balanced inverted insertions was identified. Outside of this the karyotype was normal. Genotyping microsatellite markers identified a 6 Mb deletion of all paternal alleles. This deletion was confirmed in cells with a normal karyotype, as well, using FISH. The sequence information from these genotypes allowed researchers to pinpoint a 4.82 Mb span on chromosome 1q. (Fig. 1) Ths region was known to contain ~80 genes, one which was LMNA gene. This gene drew specific attention as it is involved in six different genetic disorders, including Emery-Marie muscular dystrophy and Charcot-Marie-Tooth disease. The gene contains 12 exons and 25 kb of DNA, and codes for lamins A/C.
PCR amplification was run on all exons of the LMNA gene followed by complete sequencing. 23 samples were analyzed from patients with classical HGPS cases. 18 of the 23 samples contained identical heterozygous base substitution within exon 11, with the amino acid sequence GGC being changed to GGT. Of the five that did not contain this mutation, three were the previously discussed UPD cases (two) and the 6 Mb deletion (one). In eight of the cases parental sequence data of affected offspring showed no type mutations, thus indicating de novo mutations. The cytosine to thymine base substitution is the most mutable base in vertebrates. Additionally, one of the two sample that did not have this mutation had a different base substitution in the same codon. The amino acid sequence GGC was mutated to AGC causing a conservative substitution of serine to glycine. Further investigation into these seemingly harmless changes suggested the possibility of the activation of a cryptic splice site. Reverse transcription PCR was used to further study this possibility.
RT PCR products of unaffected individuals yield the expected product. However, individuals affected with HGPS yield a product that has a 50 amino acid deletion near the C terminus of lamin A. To check to see if this mutant messenger RNA is being translated in the affected individuals, a western blot was run. In addition to the normal lamin A and C bands, another band appears indicating the presence of a product from the mutant RNA of the 50 amino acid deletion. Of the five samples that did not contain the abnormal splice sites, 4 were discussed in the paper and possible reasons given to the phenotypes displayed. One patient had a mutation in exon 2 that caused a phenotype similar to HGPS. Upon further investigation it was found that this patient did not carry all of the characterizations on someone afflicted by classical HGPS. As for the two patient with UPD and the one with the 6 Mb deletion of all paternal alleles, it was hypothesized that these phenotypes were caused by somatic rescue events. More can be found on page 295 of the actual paper describing these hypotheses.
Much biochemical information was known about lamin A function and processing before this experiment. Erickson et al. The group hypothesized that mutations in codon 11 of HGPS patients produce a prelamin A still containing the CAAX-box, but lacks the site for endoproteolytic cleavage, two abnormalites that hinders the production of a proper mature lamin. Also it was known that phosphorylation of lamin A during the cell cycle is essential for proper lamin A maturation, and there is at least on phosphorylation site missing HGPS proteins. This type of processing of prelamin A may be dominant negative. Recent studies in mice found a gene, Zmpste 24, invovled in prelamin A processing that when knocked out a nuclear phenotype similar to HGPS patients occurs, which is abnormalities in the shape of the nuclear envelope and membrane. Immunofluorescence studies were used to further examine the structural irregularities of the nuclear membrane in unaffected parents and their affected offspring. The affected individuals showed 48% of the cells expressed the irregular nuclear shapes whereas the unaffected show less than 6% of the cells with a similar phenotype. To ensure that these differences were not artefacts from a secondary process, cell cultures from the control and HGPS lines were monitored for differences in cell cycle and apoptosis, with no significant differences found. This validated that no secondary processes occurred.
Lastly the authors of the paper noted that molecular diagnostics for HGPS could be on the market very soon after this study and that LMNA may play a large role in the natural aging process, and further studies should be conducted in this area.
Fig. 1 Summary map of candidate gene region

The figure to the left is the summary map used to identify the candidate region on chromosome 1 where the gene mutation responsible for the HGPS phenotype is present. The arrows indicate microsatellite markers and the horizontal bars show the BAC probes used for FISH. The region considered was a total of 4.82 Mb in length and represented approximately 80 genes.
Fig. 2 Point mutations in exon 11 of LMNA gene
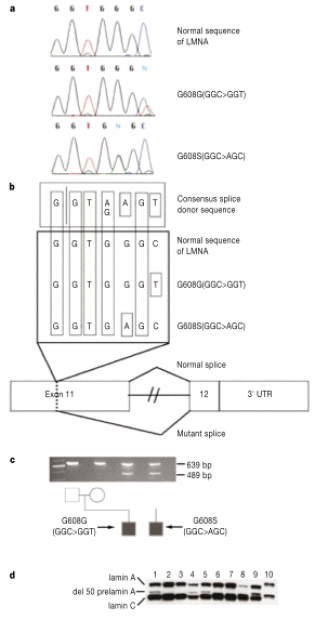
a) Sequences from one healthy individual and two patients with HGPS. b) Schematic of hypothesis for the activation of a cryptic splice site. c) RT-PCR products indicating an abnormal splice product of 489 bp. Evidence for activation of cryptic splice site. d) Western blot using antibody against lamin A/C. Lanes 1, 5, 8, 9 are from patients carrying the amino acid missense mutation GGC>GGT. Lane 4 is from a patient with amino acid missense mutation GGC>AGC. Lanes 2, 3 are from healthy parents with affected offspring. Lane 6 is from a father with an affected child and lane 7 is from a mother of an affected child. Lane 10 is a protein sample from HeLa cells.
Article reviewed and figures taken from: doi: 10/1038/nature01629.
Peter St. Andre [email protected] Last updated:3/15/09
gen677.weebly.com
